BitLocker recovery password details
Recently the BitLocker Penetration team was asked some questions about the security of the recovery password. Even if you use BitLocker every day, you may never have seen the recovery password entry screen – it is displayed by the Boot Manager in the situation where the key it needs to unlock your BitLocker protected volume is not available for some reason. A simple way to see this screen in TPM & PIN mode is to forget your PIN - you will be asked to enter a 48-digit recovery password. This password allows you to boot into Vista and set a new PIN.
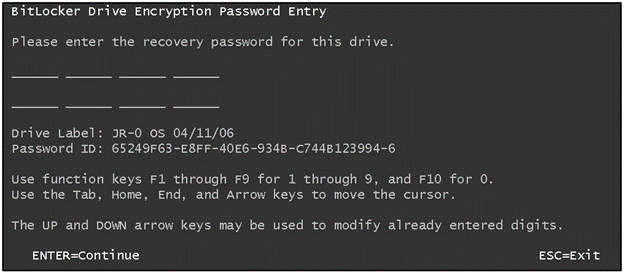
Figure 1 - Please enter 48 digits!
To help ensure that the password is entered correctly, we have you type it in in eight blocks of 6 digits. If you make a typing error while entering any block, we let you know that an error is present, and allow correction before continuing. Why do we do this? Well, manually entering 128 bits of a cryptographic key via the keyboard is not particularly easy. Users making a small error entering the key would simply be told 'incorrect key' and would have to re-enter the whole key. One of the enterprise scenarios we picture is a helpdesk technician reading a recovery key over the phone to a remote user - re-reading long keys increases support costs as well as user frustration!
The question we are being asked is 'doesn't this verification stage weaken the security?'. The answer is, as you'd hopefully expect, no - and here's why:
When we create the recovery password, we start with a random 128-bit key, which we split it into eight groups of 16 bits. Each group contains 16 bits of entropy, and can be written as a value between 0 and (2^16 - 1). We take this value and multiply it by 11. The range of values this now describes is from 0 to 11 x (2^16 - 1) (0 thru 720885). Notice that only 1 in 11 of the output are now 'valid' values. We pad with zeros, and write this as a six-digit value. This value still contains the original 16 bits of entropy, but now distributed over a larger range. We repeat the process for the other seven blocks, producing a 48 digit password.
When a user is entering the key, we accept it 6 digits at a time, and then check to see if the number they just entered is exactly divisible by 11. If it is then we know it might form part of the key - if it doesn't then we know for sure it isn't a valid block. This guards against swapped digits, mis-entered numbers, etc, and we can safely report the entry error to the user.
But does this check reduce the amount of work an attacker would have to do to brute force the underlying key? Consider that when we check a group of digits we aren't saying that they are the 'correct' group for that location in the key - merely that they could be a correct group, as they are divisible by 11. To verify this, try swapping two groups when entering your key - the individual groups will validate correctly, but the overall key will still be rejected. The work that an attacker has to do to brute-force the key is equivalent to the situation without the group-check.
Here's a small toy example, where we start with an 8-bit key (01101101). We will split it into two blocks of 4 bits:
0110 1101 = 06 13 (decimal)
we now multiply by 11:
= 66 143
Which gives us a recovery key of 6 digits [066-143]
Mistyping these blocks as 067, 606, 134, 413 etc. will result in an error - but we can't verify the whole key until we have both blocks. How much work would an attacker have to do to brute-force this key? Each block has potentially 1000 values - but the attacker knows that only those divisible by 11 can be valid keys - thus the exhaust work space is reduced to 90 values. More so, the attacker knows that all values above 165 are not valid keys (because there is no way that 4-bit value x 11 would be greater than 165), so there are only actually 16 valid values per block (0, 11, thru 165); with two blocks, this gives a key space of 16 x 16 = 256 keys to try, which is, unsurprisingly, 2^8 - or the same as the key space we began with. If the example is expanded to the full key of 48 digits the same facts about the key space hold, and the amount of work required for the attacker is not diminished.
- Jon (Penetration Test Engineer)